From Surf Wiki (app.surf) — the open knowledge base
Tyrosine aminotransferase
Mammalian protein found in Homo sapiens

Mammalian protein found in Homo sapiens
| Field | Value |
|---|---|
| Name | Tyrosine transaminase |
| EC_number | 2.6.1.5 |
| CAS_number | 9014-55-5 |
| GO_code | 0080130 |
| image | 3DYD.png |
| caption | Human tyrosine aminotransferase (rainbow colored, N-terminus = blue, C-terminus = red) complexed with pyridoxal phosphate (space-filling model). |
Tyrosine aminotransferase (or tyrosine transaminase) is an enzyme present in the liver and catalyzes the conversion of tyrosine to 4-hydroxyphenylpyruvate.
L-tyrosine + 2-oxoglutarate \rightleftharpoons 4-hydroxyphenylpyruvate + L-glutamate
In humans, the tyrosine aminotransferase protein is encoded by the TAT gene. A deficiency of the enzyme in humans can result in what is known as type II tyrosinemia, wherein there is an abundance of tyrosine as a result of tyrosine failing to undergo an aminotransferase reaction to form 4-hydroxyphenylpyruvate.
Function
Tyrosine aminotransferase (TAT) is a pyridoxal phosphate (PLP)-dependent enzyme that catalyzes the first and rate-limiting step in the degradation of the amino acid tyrosine, transferring its amino group to α-ketoglutarate to produce 4-hydroxyphenylpyruvate and glutamate. This reaction is essential for tyrosine catabolism and energy production, as the resulting products feed into the citric acid cycle. TAT is predominantly expressed in the liver but is also present in other tissues, where it maintains narrow substrate specificity, favoring tyrosine over other aromatic amino acids such as phenylalanine. Structurally, TAT contains a central catalytic core and a PLP-binding site critical for its enzymatic function.
Structure

Tyrosine aminotransferase is a dimeric enzyme composed of two identical subunits, each containing a central catalytic core and a smaller domain formed by both amino- and carboxyl-terminal regions. The enzyme's active site features a lysine residue (Lys280 in humans) that forms a Schiff base with the pyridoxal phosphate (PLP) cofactor, which is essential for its transaminase activity. Structural studies, including crystallography, reveal that the PLP cofactor is stabilized within the active site through interactions with nonpolar amino acid side chains and hydrogen bonding, facilitating precise substrate positioning and catalysis. Additionally, the carboxyl terminus contains hydrophilic, glutamate-rich segments resembling PEST sequences, which may contribute to the enzyme's rapid degradation rate. The overall architecture of tyrosine aminotransferase aligns it with the aminotransferase superfamily, sharing conserved residues critical for function and substrate specificity.
Mechanism
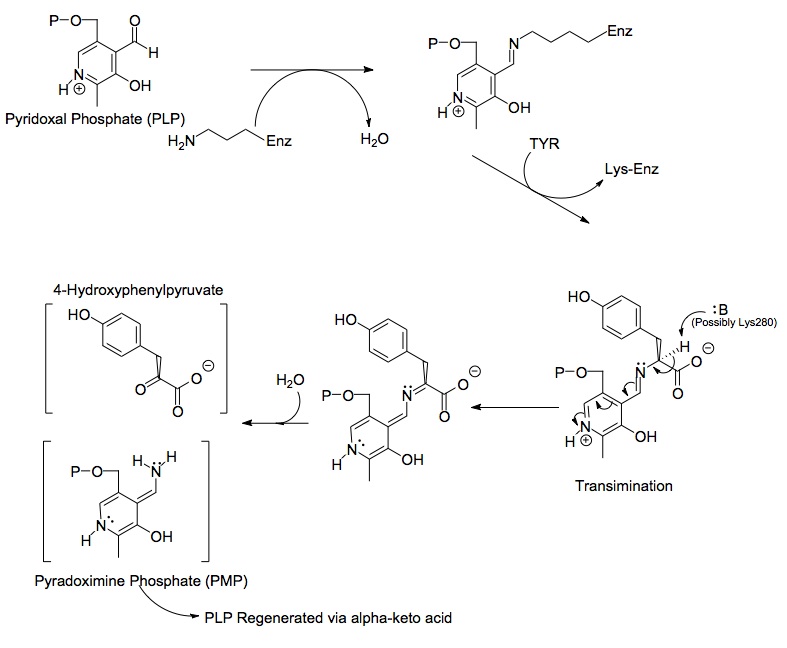
Structures of the three main molecules involved in chemical reaction catalyzed by the tyrosine aminotransferase enzyme are shown below: the amino acid tyrosine (left), the prosthetic group pyridoxal phosphate (right), and the resulting product 4-hydroxyphenylpyruvate (center).
Each side of the dimer protein includes pyridoxal phosphate (PLP) bonded to the Lys280 residue of the tyrosine aminotransferase molecule. The amine group of tyrosine attacks the alpha carbon of the imine bonded to Lys280, forming a tetrahedral complex and then kicking off the LYS-ENZ. This process is known as transimination by the act of switching out the imine group bonded to PLP. The newly formed PLP-TYR molecule is then attacked by a base.
A possible candidate for the base in the mechanism could be Lys280 that was just pushed off of PLP, which sequesters the newly formed amino group of the PLP-TYR molecule. In a similar mechanism of aspartate transaminase, the lysine that forms the initial imine to PLP later acts as the base that attacks the tyrosine in transimination. The electrons left behind from the loss of the proton move down to form a new double bond to the imine, which in turn pushes the already double bonded electrons through PLP and end up as a lone pair on the positively charged nitrogen in the six-membered ring of the molecule. Water attacks the alpha carbon of the imine of PLP-TYR and through acyl substitution kicks off the nitrogen of PLP and forming pyridoxamine phosphate (PMP) and 4-hydroxyphenylpyruvate.
PMP is then regenerated into PLP by transferring its amine group to alpha-ketoglutarate, reforming its aldehyde functional group. This is followed by another substitution reaction with the Lys280 residue to reform its imine linkage to the enzyme, forming ENZ-PLP.
Clinical significance
Deficiency of TAT activity leads to tyrosinemia type II, a metabolic disorder marked by elevated blood tyrosine levels and symptoms including keratitis, skin lesions, and neurological impairment.
Tyrosinemia is the most common metabolic disease associated with tyrosine aminotransferase. The disease results from a deficiency in hepatic tyrosine aminotransferase. Tyrosinemia type II (Richner-Hanhart syndrome, RHS) is a disease of autosomal recessive inheritance characterized by keratitis, palmoplantar hyperkeratosis, mental retardation, and elevated blood tyrosine levels. Keratitis in Tyrosinemia type II patients is caused by the deposition of tyrosine crystals in the cornea and results in corneal inflammation. The TAT gene is located on human chromosome 16q22-24 and extends over 10.9 kilobases (kb) containing 12 exons, and its 3.0 kb mRNA codes for a 454-amino acid protein of 50.4 kDa. Twelve different TAT gene mutations have been reported.
References
References
- (2008). "Human tyrosine aminotransferase". Protein Data Bank.
- (April 1992). "Tyrosine aminotransferase: a transaminase among others?". Cellular and Molecular Biology.
- (27 November 2017). "The Incidence of Transient Neonatal Tyrosinemia Within a Mexican Population". Journal of Inborn Errors of Metabolism and Screening.
- (July 1990). "Isolation and characterization of the human tyrosine aminotransferase gene". Nucleic Acids Research.
- (May 2006). "The narrow substrate specificity of human tyrosine aminotransferase--the enzyme deficient in tyrosinemia type II". The FEBS Journal.
- (July 2019). "Analysis of Chromatin Binding of Ectopically Expressed Proteins in Early Xenopus Embryos". Cold Spring Harbor Protocols.
- (January 1989). "The structure of tyrosine aminotransferase. Evidence for domains involved in catalysis and enzyme turnover". The Journal of Biological Chemistry.
- (October 2004). "UCSF Chimera--a visualization system for exploratory research and analysis". Journal of Computational Chemistry.
- (June 2011). "The beneficial effects of α-cyclodextrin on blood lipids and weight loss in healthy humans". Obesity.
- (May 2006). "The genetic tyrosinemias". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics.
- (October 1992). "Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II". Proceedings of the National Academy of Sciences of the United States of America.
- (March 1995). "Richner-Hanhart syndrome (tyrosinemia type II). Case report and literature review". Ophthalmic Genetics.
- (January 2006). "Richner-Hanhart syndrome: report of a case with a novel mutation of tyrosine aminotransferase". Journal of Dermatological Science.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Tyrosine aminotransferase — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report