From Surf Wiki (app.surf) — the open knowledge base
Discovery and development of direct thrombin inhibitors
Drug discovery

Drug discovery
Direct thrombin inhibitors (DTIs) are a class of anticoagulant drugs that can be used to prevent and treat embolisms and blood clots caused by various diseases. They inhibit thrombin, a serine protease which affects the coagulation cascade in many ways. DTIs have undergone rapid development since the 90's. With technological advances in genetic engineering the production of recombinant hirudin was made possible which opened the door to this new group of drugs. Before the use of DTIs the therapy and prophylaxis for anticoagulation had stayed the same for over 50 years with the use of heparin derivatives and warfarin which have some well known disadvantages. DTIs are still under development, but the research focus has shifted towards factor Xa inhibitors, or even dual thrombin and fXa inhibitors that have a broader mechanism of action by both inhibiting factor IIa (thrombin) and Xa. A recent review of patents and literature on thrombin inhibitors has demonstrated that the development of allosteric and multi-mechanism inhibitors might lead the way to a safer anticoagulant.
History
Anticoagulation therapy has a long history. In 1884 John Berry Haycraft described a substance found in the saliva of leeches, Hirudo medicinalis, that had anticoagulant effects. He named the substance ‘Hirudine’ from the Latin name. The use of medicinal leeches can be dated back all the way to ancient Egypt. In the early 20th century Jay McLean, L. Emmet Holt Jr. and William Henry Howell discovered the anticoagulant heparin, which they isolated from the liver (hepar). Heparin remains one of the most effective anticoagulants and is still used today, although it has its disadvantages, such as requiring intravenous administration and having a variable dose-response curve due to substantial protein binding. In the 1980s low molecular-weight heparin (LMWH) were developed. They are derived from heparin by enzymatic or chemical depolymerization and have better pharmacokinetic properties than heparin. In 1955 the first clinical use of warfarin, a vitamin K antagonist, was reported. Warfarin was originally used as a rat poison in 1948 and thought to be unsafe for humans, but a suicide attempt suggested that it was relatively safe for humans. Vitamin K antagonists are the most commonly used oral anticoagulants today and warfarin was the 11th most prescribed drug in the United States in 1999 Warfarin has its disadvantages though, just like heparin, such as a narrow therapeutic index and multiple food and drug interactions and it requires routine anticoagulation monitoring and dose adjustment. Since both heparin and warfarin have their downsides the search for alternative anticoagulants has been ongoing and DTIs are proving to be worthy competitors. The first DTI was actually hirudin, which became more easily available with genetic engineering. It is now available in a recombinant form as lepirudin (Refludan) and desirudin (Revasc, Iprivask). Development of other DTIs followed with the hirudin analog, bivalirudin, and then the small molecular DTIs. However, such DTIs were also having side effects such as bleeding complications and liver toxicity, and their long-term effects were in doubt.
Mechanism of action
Blood clotting cascade

When a blood vessel ruptures or gets injured, factor VII comes into contact with tissue factors which starts a process called the blood coagulation cascade. Its purpose is to stop bleeding and repair tissue damage. When this process is too active due to various problems the risk of blood clots or embolisms increases. As the name indicates the cascade is a multi-step procedure where the main product thrombin is made by activating various proenzymes (mainly serine proteases) in each step of the cascade. Thrombin has multiple purposes, but mainly it converts soluble fibrinogen to an insoluble fibrin complex. Furthermore, it activates factors V, VIII and XI, all by cleaving the sequences GlyGlyGlyValArg-GlyPro and PhePheSerAlaArg-GlyHis, selectively between Arginine (Arg) and Glycine (Gly). These factors generate more thrombin. Thrombin also activates factor XIII that stabilizes the fibrin complex and therefore the clot and it stimulates platelets, which help with the coagulation. Given this broad action of thrombin it stands as a good drug target for anticoagulant drugs such as heparin, warfarin and DTIs and antiplatelet drugs like aspirin.
Binding sites
Thrombin is in the serine protease family. It has 3 binding domains in which thrombin-inhibition drugs bind to. Those proteases have a deep narrow gap as an active binding site that consists of two β-barrel subdomains that make up the surface gap which binds substrate peptides. The surface in the gap seems to have limiting access to molecules by steric hindrance, this binding site consists of 3 amino acids, Asp-102, His-57 and Ser-195. Thrombin also has two exosites (1 and 2). Thrombin is a little different from other serine proteases as exosite 1 is anion-binding and binds to fibrin and other similar substrates while exosite 2 is a heparin-binding domain.
DTIs inhibition
DTIs inhibit thrombin by two ways; bivalent DTIs block simultaneously the active site and exosite 1 and act as competitive inhibitors of fibrin, while univalent DTIs block only the active site and can therefore both inhibit unbound and fibrin-bound thrombin. In contrast, heparin drugs bind in exosite 2 and form a bridge between thrombin and antithrombin, a natural anticoagulant substrate formed in the body, and strongly catalyzes its function. But heparin can also form a bridge between thrombin and fibrin which binds to exosite 1 which protects the thrombin from inhibiting function of heparin-antithrombin complex and increases thrombin's affinity to fibrin. DTIs that bind to the anion-binding site have shown to inactivate thrombin without disconnecting thrombin from fibrin, which points to a separate binding site for fibrin. DTIs aren't dependent to cofactors like antithrombin to inhibit thrombin so they can both inhibit free/soluble thrombin as well as fibrin bound thrombin unlike heparins. The inhibition is either irreversible or reversible. Reversible inhibition is often linked to lesser risk of bleeding. Due to this action of DTIs they can both be used for prophylaxis as well as treatment for embolisms/clots.
Active site's pockets
DTIs that fit in the active binding site have to fit in the hydrophobic pocket (S1) that contains aspartic acid residue at the bottom which recognizes the basic side chain. The S2 site has a loop around tryptophan which occludes a hydrophobic pocket that can recognize larger aliphatic residues. The S3 site is flat and the S4 site is hydrophobic, it has tryptophan lined by leucine and isoleucine.
Nα-(2-naphthyl-sulphonyl-glycyl)-DL-p-amidinophenylalanyl-piperidine (NAPAP) binds thrombin in the S1, S2 and S4 pockets. The amidine group on NAPAP forms a bidentate salt bridge with Asp deep in the S1 pocket, the piperidine group takes the role of proline residue and binds in the S2 pocket, and the naphthyl rings of the molecule forms a hydrophobic interaction with Trp in the S4 pocket. Pharmaceutical companies have used the structural knowledge of NAPAP to develop DTIs. Dabigatran, like NAPAP binds to S1, S2 and S4 pockets. Benzamidine group on the dabigatran structure binds deep in the S1 pocket, the methylbenzimidazole fits nicely in the hydrophobic S2 pocket and the Ile and Leu at the bottom of the S4 pocket binds to the aromatic group of dabigatran.
Drug development
Hirudin derivatives
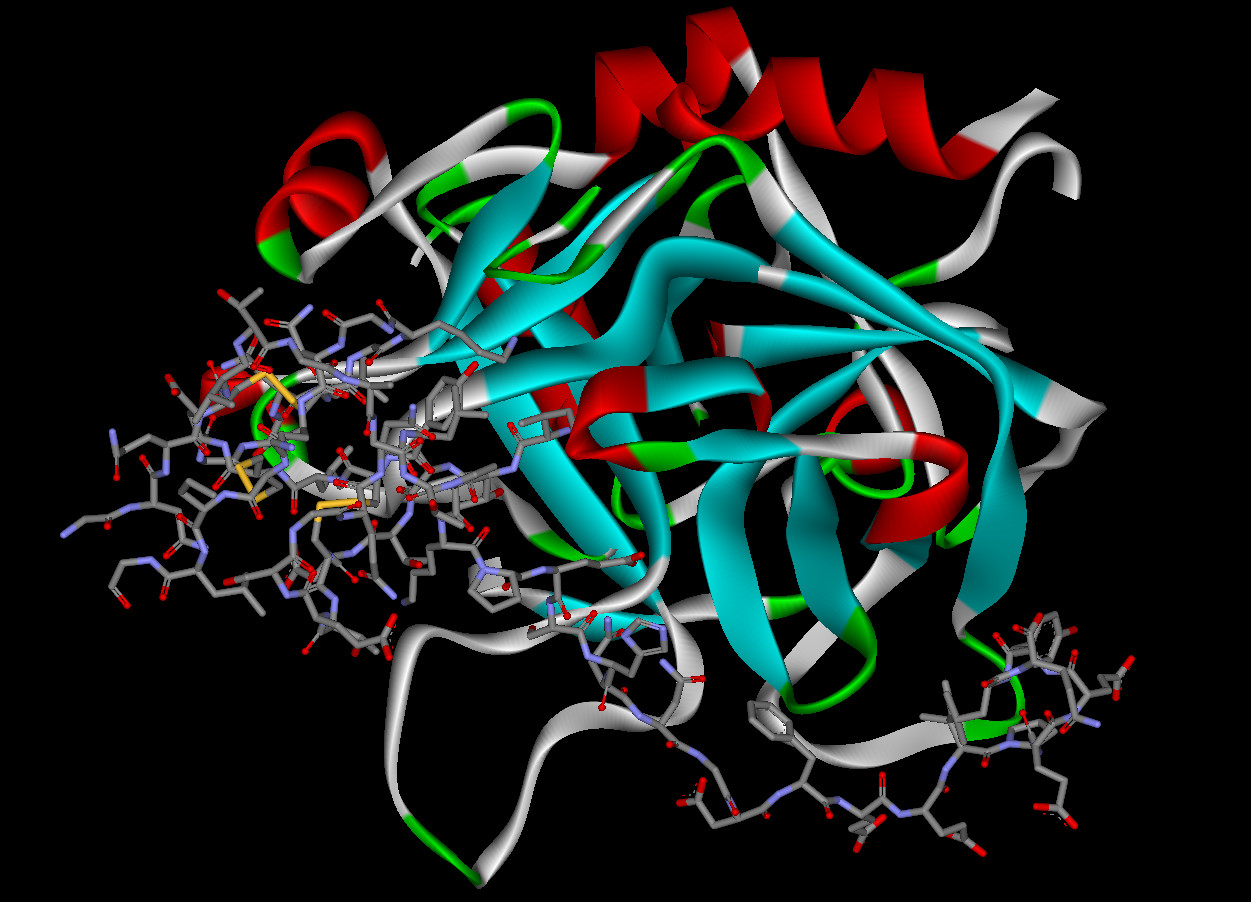
Hirudin derivatives are all bivalent DTIs, they block both the active site and exosite 1 in an irreversible 1:1 stoichiometric complex. Native hirudin, a 65-amino-acid polypeptide, is produced in the parapharyngeal glands of medicinal leeches. Hirudins today are produced by recombinant biotechnology using yeast. These recombinant hirudins lack a sulfate group at Tyr-63 and are therefore called desulfatohirudins. They have a 10-fold lower binding affinity to thrombin compared to native hirudin, but remain a highly specific inhibitor of thrombin and have an inhibition constant for thrombin in the picomolar range. Renal clearance and degradation account for the most part for the systemic clearance of desulfatohirudins and there is accumulation of the drug in patients with chronic kidney disease. These drugs should not be used in patients with impaired renal function, since there is no specific antidote available to reverse the effects. Hirudins are given parenterally, usually by intravenous injection. 80% of hirudin is distributed in the extravascular compartment and only 20% is found in the plasma. The most common desulfatohirudins today are lepirudin and desirudin.
Hirudin
In a meta-analysis of 11 randomized trials involving hirudin and other DTIs versus heparin in the treatment of acute coronary syndrome (ACS) it was found that hirudin has a significantly higher incidence of bleeding compared with heparin. Hirudin is therefore not recommended for treatment of ACS and currently it has no clinical indications.
Lepirudin

Lepirudin is approved for the treatment of heparin-induced thrombocytopenia (HIT) in the USA, Canada, Europe and Australia. HIT is a very serious adverse event related to heparin and occurs with both unfractionated heparin and LMWH, although to a lesser extent with the latter. It is an immune-mediated, prothrombotic complication which results from a platelet-activating immune response triggered by the interaction of heparin with platelet factor 4 (PF4). The PF4-heparin complex can activate platelets and may cause venous and arterial thrombosis. When lepirudin binds to thrombin it hinders its prothrombotic activity. Three prospective studies, called the Heparin-Associated-Thrombocytopenia (HAT) 1,2, and 3, were performed that compared lepirudin with historical controls in the treatment of HIT. All three studies showed that the risk of new thrombosis was decreased with the use of lepirudin, but the risk for major bleeding was increased. The higher incidence of major bleeding is thought to be the result of an approved dosing regimen that was too high, consequently the recommended dose was halved from the initial dose. As of May 2012 Bayer HealthCare, the only manufacturer of lepirudin injections, discontinued its production. They expect supplies from wholesalers to be depleted by mid-2013.
Desirudin
Desirudin is approved for treatment of venous thromboembolism (VTE) in Europe and multiple phase III trials are presently ongoing in the USA. Two studies comparing desirudin with enoxaparin (a LMWH) or unfractionated heparin have been performed. In both studies desirudin was considered to be superior in preventing VTE. Desirudin also reduced the rate of proximal deep vein thrombosis. Bleeding rates were similar with desirudin and heparin.
Bivalirudin

Bivalirudin, a 20 amino acid polypeptide, is a synthetic analog of hirudin. Like the hirudins it is also a bivalent DTI. It has an amino-terminal D-Phe-Pro-Arg-Pro domain that is linked via four Gly residues to a dodecapeptide analog of the carboxy-terminal of hirudin. The amino-terminal domain binds to the active site and the carboxy-terminal domain binds to exosite 1 on thrombin. Different from the hirudins, once bound thrombin cleaves the Arg-Pro bond at the amino-terminal of bivalirudin and as a result restores the functions to the active site of the enzyme. Even though the carboxy-terminal domain of bivalirudin is still bound to exosite 1 on thrombin, the affinity of the bond is decreased after the amino-terminal is released. This allows substrates to substrates to compete with cleaved bivalirudin for access to exosite 1 on thrombin. The use of bivalirudin has mostly been studied in the setting of acute coronary syndrome. A few studies indicate that bivalirudin is non-inferior compared to heparin and that bivalirudin is associated with a lower rate of bleeding. Unlike the hirudins, bivalirudin is only partially (about 20%) excreted by the kidneys, other sites such as hepatic metabolism and proteolysis also contribute to its metabolism, making it safer to use in patients with renal impairment; however, dose adjustments are needed in severe renal impairment.
Small molecular direct thrombin inhibitors
Small molecular direct thrombin inhibitors (smDTIs) are non-peptide small molecules that specifically and reversibly inhibit both free and clot-bound thrombin by binding to the active site of the thrombin molecule. They prevent VTE in patients undergoing hip- and knee replacement surgery.
The smDTIs where derived using a peptidomimetic design with either P1 residue from arginine itself (e.g. argatroban) or arginine-like substrates such as benzamidine (e.g. NAPAP).
Argatroban

Argatroban is a small univalent DTI formed from P1 residue from arginine. It binds to the active site on thrombin. The plasma half-life is approximately 45 minutes. As argatroban is metabolized via hepatic pathway and is mainly excreted through the biliary system, dose adjustments are necessary in patients with hepatic impairment but not renal damage. Argatroban has been approved in the USA since 2000 for the treatment of thrombosis in patients with HIT and 2002 for anticoagulation in patients with a history of HIT or are at risk of HIT undergoing percutaneous coronary interventions (PCI). It was first introduced in Japan in 1990 for treatment of peripheral vascular disorders.
Ximelagatran
The publication of the NAPAP-fIIa crystal structure triggered many researches on thrombin inhibitors. NAPAP is an active site thrombin inhibitor. It fills the S3 and S2 pockets with its naphthalene and piperidine groups. AstraZeneca used the information to develop melagatran. The compound was poorly orally available, but after renovation they got a double prodrug which was the first oral DTI in clinical trials, ximelagatran.
Dabigatran etexilate
Researchers at Boehringer Ingelheim also used the publicized information about the NAPAP-fIIa crystal structure, starting with the NAPAP structure that led to the discovery of dabigatran, a highly lipophilic, gastrointestinally absorbed and orally bioavailable double prodrug such as ximelagatran, with the plasma half-life of approximately 12 hours. Dabigatran etexilate is rapidly absorbed, it lacks interaction with cytochrome P450 enzymes and with other food and drugs, there is no need for routine monitoring and it has a broad therapeutic index and a fixed-dose administration, which is excellent safety compared with warfarin. Unlike ximelagatran, a long-term treatment of dabigatran etexilate has not been linked with hepatic toxicity, seeing as how the drug is predominantly eliminated (80%) by the kidneys. Dabigatran etexilate was approved in Canada and Europe in 2008 for the prevention of VTE in patients undergoing hip- and knee surgery. In October 2010 the US FDA approved dabigatran etexilate for the prevention of stroke in patients with atrial fibrillation (AF). Many pharmaceutical companies have attempted to develop orally bioavailable DTI drugs but dabigatran etexilate is the only one to reach the market.
In a 2012 meta-analysis dabigatran was associated with increased risk of myocardial infarction (MI) or ACS when tested against different controls in a broad spectrum of patients.
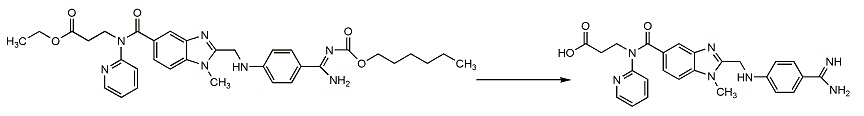
Table 1: Summary of characteristics of DTIs
| Dabigatran etexilate | U | Oral | Primarily renal, remainder is conjugated with glucuronic acid in liver | Reversible | Prevention of stroke and embolism in patients with AF | Rapid onset of action, lack of interaction with CYP450, food or drugs, broad TI, fixed dose administration and good safety profile, not associated with hepatotoxicity for long-term use |
|---|
iv: intravenous, sc: subcutaneous, HIT: heparin-induced thrombocytopenia, VTE: Venous thromboembolism, DVT: Deep vein thrombosis, PTCA: Percutaneous transluminal coronary angioplasty, PCI: percutaneous coronary intervention, FDA: Food and Drug Administration, AF: Atrial fibrillation, TI: Therapeutic index
Status 2014
In 2014 dabigatran remains the only approved oral DTI which is a follow-up compound of ximelgatran that is being developed by AstraZeneca. It is the prodrug of a potent, competitive, reversible inhibitor of free and fibrin-bound thrombin called ARH0637. In a Phase 2 trial for AF the mean serum creatinine concentration increased by about 10% from baseline in patients treated with AZD0837, which returned to baseline after cessation of therapy. Development of other oral DTIs, such as Sofigatran from Mitsubishi Tanabe Pharma has been discontinued. Another strategy for developing oral anticoagulant drugs is that of dual thrombin and fXa inhibitors that some pharmaceutical companies, including Boehringer Ingelheim, have reported on. These compounds show favorable anticoagulant activity in vitro.
References
References
- Mehta, AY. (Jan 2014). "An update on recent patents on thrombin inhibitors (2010 - 2013).". Expert Opinion on Therapeutic Patents.
- Whitacker, I.S.. (2004). "Historical Article: Hirudo medicinalis: ancient origins of, and trends in the use of medicinal leeches throughout history". British Journal of Oral and Maxillofacial Surgery.
- Shapiro, Sandor S.. (2003). "Treating Thrombosis in the 21st Century". New England Journal of Medicine.
- O’Brien, P. Joshua. (2012). "Direct Thrombin Inhibitors". Journal of Cardiovascular Pharmacology and Therapeutics.
- Hirsh, Jack. (2004). "Heparin and Low-Molecular-Weight Heparin The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy". Chest.
- Thethi, Indermohan. (1 January 2012). "Oral Factor Xa and Direct Thrombin Inhibitors". Journal of Burn Care & Research.
- Kendoff, D.. (30 December 2011). "Oral Thromboprophylaxis Following Total Hip or Knee Replacement: Review and Multicentre Experience with Dabigatran Etexilate". The Open Orthopaedics Journal.
- Di Nisio, Marcello J.. (2005). "Direct Thrombin Inhibitors". New England Journal of Medicine.
- Nar, Herbert. (2012). "The role of structural information in the discovery of direct thrombin and factor Xa inhibitors". Trends in Pharmacological Sciences.
- Lee, Catherine J.. (2011). "Direct Thrombin Inhibitors". British Journal of Clinical Pharmacology.
- Patrono, C. (May 5, 1994). "Aspirin as an antiplatelet drug". New England Journal of Medicine.
- Lefkovits, J.. (1994). "Direct thrombin inhibitors in cardiovascular medicine.". Circulation.
- Thomas, edited by Simon Redwood, Nick Curzen, Martyn R.. (2010). "Oxford textbook of interventional cardiology". Oxford University Press.
- Greinacher, Andreas. (2008). "The direct thrombin inhibitor hirudin". Thrombosis and Haemostasis.
- Sakr, Yasser. (2011). "Heparin-induced thrombocytopenia in the ICU: an overview". Critical Care.
- "Lepirudin Injection". American Society of Health-System Pharmacists.
- Squizzato, A. (2009). "New direct thrombin inhibitors". Intern Emerg Med.
- Uchino, K.. (9 January 2012). "Dabigatran Association With Higher Risk of Acute Coronary Events: Meta-analysis of Noninferiority Randomized Controlled Trials". Archives of Internal Medicine.
- "Angiomax Injection".
- "FDA approves Pradaxa to prevent stroke in people with atrial fibrillation".
- "AZD0837". Astrazenecaclinicaltrials.com.
- AstraZeneca Long-term treatment with the oral direct thrombin inhibitor AZD0837, compared to Vitamin-K antagonists, as stroke prevention in patients with non-valvular atrial fibrillation and one or more risk factors for stroke and systemic embolic events. A 5-year follow-up study study code D1250C0004221 January 2010 [http://www.astrazenecaclinicaltrials.com/_mshost800325/content/clinical-trials/resources/pdf/D1250C00042 Trial D1250C00042] {{webarchive. link. (November 10, 2013)
- (2010). "Update on Antithrombotic Therapy: New Anticoagulants". Circulation.
- (December 2009). "Oral direct thrombin inhibitor AZD0837 for the prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation: a randomized dose-guiding, safety, and tolerability study of four doses of AZD0837 vs. vitamin K antagonists". Eur. Heart J..
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Discovery and development of direct thrombin inhibitors — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report